Entering the rapidly growing digital health industry is exciting and promising for established and entrepreneurial companies alike. While exciting, navigating the complicated regulatory structures of different countries is one of the first hurdles any established or emerging digital health company needs to overcome.
Healthcare regulatory bodies across the globe are adapting and expanding their existing processes to ensure proven digital health solutions can be quickly implemented into clinical settings. Several countries have, for instance, established fast-track programs to expedite the regulatory approval of DTx products, i.e. digital health solutions that treat, manage or prevent a disease, are backed by clinical evidence and real-world outcomes and require regulatory review. Learn more about DTx in our DTx 101 Series, Introducing Digital Therapeutics.
Several countries in the European Union (EU), such as Germany, France, and Belgium, are defining regulatory steps to integrate DTx products into their healthcare systems. The fragmented nature of the EU regulatory landscape can, however, make it challenging for European digital health companies to scale their organization successfully and rapidly. For those companies, the United States (US) can be an enticing market for both regulatory and commercial reasons. First of all, the FDA serves as a single regulatory body for a population as large as 330 M inhabitants, exceeding the population size of Western and Northern Europe combined. Besides that, the regulatory pathways are longer established than elsewhere in the world and reimbursement options are growing, providing DTx companies with concrete handholds and learnings on how to enter this sizeable market.
Below we outline the different regulatory models used in the EU, and explore why the US can be seen as an interesting geographical market for DTx solutions.
Germany
The German digital health app pathway (DiGA) sets the standard for European DTx integration. Recent legislation embedded DiGA into Germany’s Federal Institute for Drugs and Medical Devices (BfArM), therefore, enabling the reimbursement of DTx solutions. On a high-level to sell DTx products in Germany, DTx companies must:
- Acquire a conformity mark (CE mark) from the EU prior to initiating the DiGA approval process;
- Following the CE mark, the DTx product is evaluated as part of DiGA for scientific merit, safety, user interface, and other considerations;
- If all requirements are met, the DTx product achieves a permanent DiGA listing, allowing physician prescription and reimbursement by state insurance;
- Reimbursement rate is determined by the German National Association of Statutory Health Insurance Funds (GKV-SV), a semi-governmental organization with parallels to the governing body issuing US Current Procedural Terminology Codes (CPT codes).
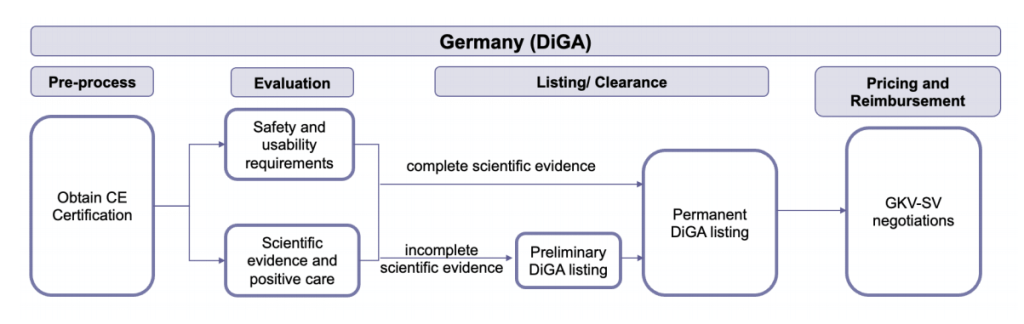
Figure 1: Germany’s DiGA process: DTx products undertake this pathway to secure regulatory approval and obtain a pricing and reimbursement arrangement within the German health system.
France and Belgium
France and Belgium have both developed dedicated pathways for DTx regulation. France follows the German example of a fast-track process to make DTx products available and reimbursable through their public health system. In comparison, Belgium has introduced a validation pyramid with three levels to determine whether a DTx can be reimbursed.
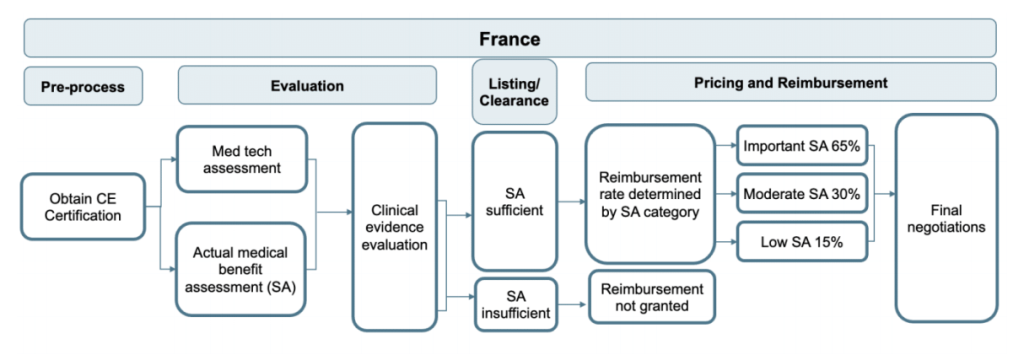
Figure 2: France’s regulatory pathway for DTx products
Compared to the German approach, the French and Belgian systems require more stringent proof of a DTx product’s ability to add socio-economic value prior to approving reimbursement. For example, the Belgian DTx company moveUP initiated a clinical study dedicated to proving the socio-economic impact of their app that provides personalized exercise plans for patients that underwent a joint replacement. According to an earlier news article, the company claims that it can generate €27M annual savings for the Belgian health insurance administrator INAMI.1
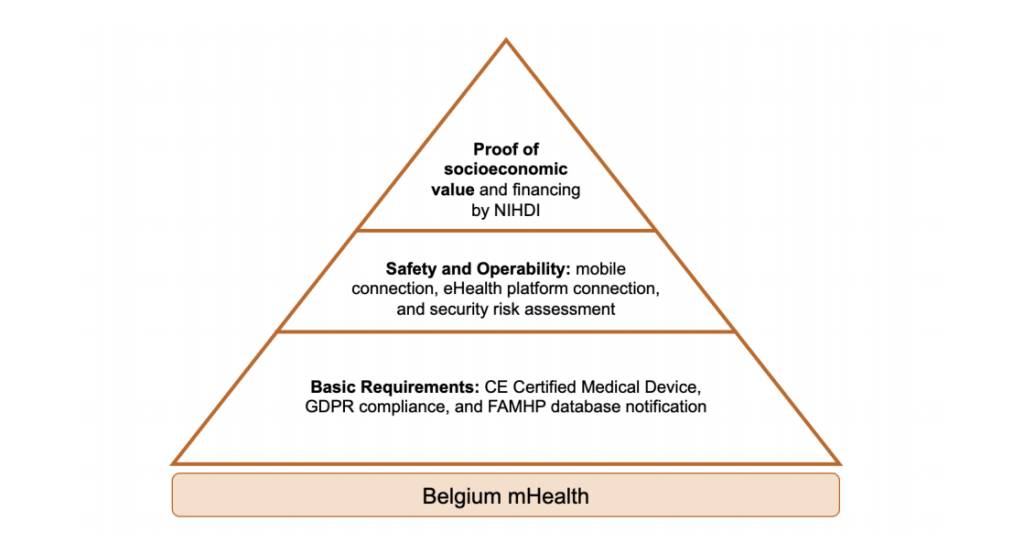
Figure 3: Belgium’s regulatory pathway for DTx products. (Abbreviations: CE- Conformity Mark, GDPR- General Data Protection Regulation, FAMHP- Federal Agency for Medicines and Health Products, NIHDI- National Institute for Health and Disability Insurance)
United States
Regulation
While the European countries mentioned above built and piloted new pathways for DTx regulation, the US has integrated DTx regulation into existing Food and Drug Administration (FDA) pathways traditionally used for medical devices. Unlike other countries, pulling DTx into existing pathways allows digital solutions to be less differentiated from the traditional regulation of devices and drugs in use by providers and current reimbursement approaches.
Most commonly, DTx products enter the FDA regulatory process through the 510(k) or De Novo avenues. Some DTx products meeting more stringent criteria can gain approval through the Breakthrough Device Designation pathway. Other lesser-trodden FDA pathways, such as STeP, can apply to DTx products under special circumstances. Learn more about FDA regulation for DTx in our blog Decimal.reg: Let Us Guide You Through the FDA Approval Process.
The US FDA pathways, while more nuanced than their European counterparts, have potential for greater DTx success. The DTx, NightWare, indicated for treatment of PTSD-induced nightmares or nightmare disorder, exemplifies this success. First designated as a Breakthrough Device, NightWare moved forward to FDA clearance through the De Novo pathway and has since raised triple its pre-clearance funding and is making significant strides in revenue. A clear strategy for navigating these FDA pathways is crucial for success and minimizing the time and cost of undergoing this process.

Figure 3: Overview of the FDA’s regulatory pathways for DTx products. These relevant paths as well as other paths and entry points are not mutually exclusive
Financial Implications
Overall, the US is a financially lucrative market to enter. In Germany, France, and Belgium, pricing is strictly evaluated and controlled by governmental agencies to obtain a low price for both the government and the patient. Unlike those countries, the US doesn’t operate under a universal health system model. This lesser degree of government involvement in reimbursement sets the stage for more lucrative DTx entry.
In the US, reimbursement for DTx products is usually determined by a CPT code acquired from an independent organization that is separate from the FDA. Depending on your company’s target customer segment, there are alternative ways to offer a DTx product to patients without a CPT code. For more insight into US reimbursement see our blog, Tackling Digital Therapeutic Reimbursement.
Additionally, CPT codes do not always completely account for the final pricing of a therapeutic or intervention. Different payers (such as Medicare, Medicaid or various private insurance companies) and providers that prescribe the DTx and bill with the CPT code may have additional, variable costs. This means the same healthcare product almost always sells for a higher price in the US than other countries, usually by at least 50%.2
Takeaways
The US’s regulatory pathways for DTx offer greater opportunity for market entry due to many existing avenues for regulation and multiple possibilities concerning reimbursement and payment. Compared to the individual EU markets, the US market itself is larger in terms of number of reachable patients under a single regulatory body as well as reimbursement amount that can be attained.
While worth the reward, navigating the US systems for regulation and reimbursement is complicated. As experts in this navigation, Decimal.health would be honored to serve as your partner in navigating DTx regulation and reimbursement to maximize your success in the US.

